L’ammoniac (NH3), une molécule bien connue aux multiples applications, suscite un regain d’intérêt en tant que potentielle ressource énergétique renouvelable. Des chercheurs explorent actuellement de nouvelles technologies liées à l’ammoniac, nécessitant des données précises sur ses propriétés thermodynamiques fondamentales. Le Dr Jadran Vrabec et son équipe ont récemment publié les résultats de simulations de pointe, ouvrant le chemin à de nouvelles applications de l’ammoniac.
Produit final du célèbre procédé Haber-Bosch, l’ammoniac est couramment synthétisé pour capter l’azote destiné aux engrais. Il est également utilisé dans la réfrigération, les produits d’entretien et la production de médicaments. Stable et sûr à manipuler, combustible et contenant la plus grande fraction d’hydrogène de toutes les molécules à l’exception de l’hydrogène pur, l’ammoniac pourrait constituer une alternative viable aux vecteurs énergétiques à base de carbone responsables du changement climatique.
Des recherches explorent déjà comment l’ammoniac pourrait alimenter directement des moteurs, des turbines à gaz et des piles à hydrogène. Il pourrait aussi servir à stocker de l’énergie lorsque d’autres sources renouvelables comme l’éolien et le solaire ne peuvent répondre à la demande.
Le besoin de nouvelles données thermodynamiques
Si l’ammoniac est bien connu, l’intérêt pour son utilisation comme carburant a initié une quête de nouvelles technologies et un besoin accru de données précises sur ses propriétés thermodynamiques fondamentales parmi les ingénieurs chimistes. Ces propriétés, comme les équilibres de phase, la densité ou la capacité thermique, caractérisent les systèmes physiques et déterminent le fonctionnement des processus chimiques.
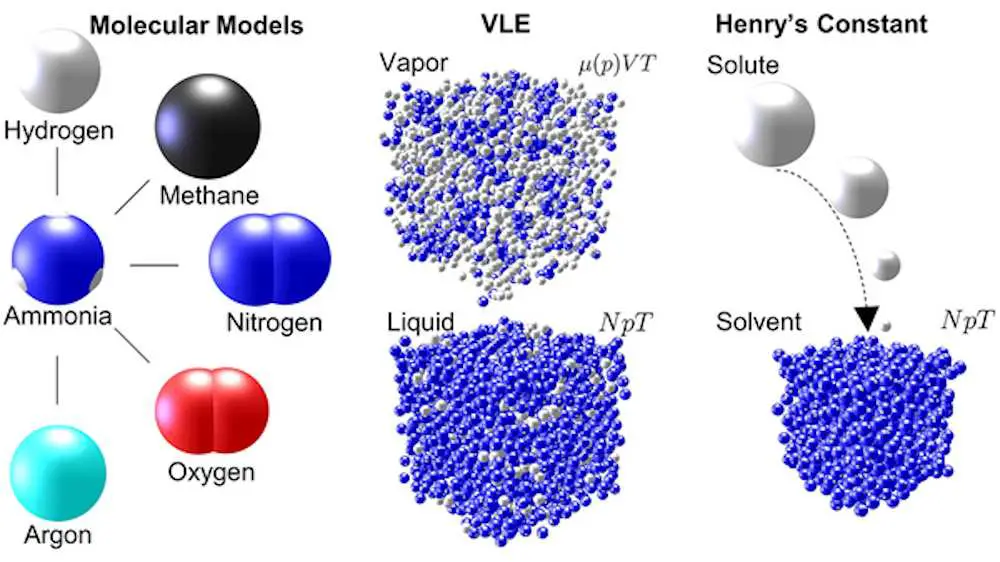
Les ingénieurs aimeraient aussi mieux comprendre comment ces propriétés évoluent lorsque l’ammoniac est mélangé à d’autres molécules, afin d’optimiser les processus et les conditions opératoires. Selon le Dr Vrabec, directeur de l’Institut des sciences des procédés de l’Université technique de Berlin, « les propriétés thermodynamiques sont à 100% déterminées par les interactions moléculaires » qui ne peuvent être étudiées qu’en réalisant de grandes simulations sur des supercalculateurs.
Des simulations à grande échelle sur les propriétés de l’ammoniac
Dans un article récent publié dans le Journal of Chemical & Engineering Data, le Dr Vrabec et Erich Mace rapportent les résultats de simulations réalisées sur le supercalculateur Hawk du High-Performance Computing Center Stuttgart (HLRS), se concentrant sur les propriétés thermodynamiques de mélanges contenant de l’ammoniac.
Leur approche s’appuie sur les concepts de thermodynamique énoncés par Ludwig Boltzmann au 19e siècle, devenus applicables dans les années 1950 avec l’arrivée des premiers ordinateurs. Depuis, le domaine a progressé parallèlement au développement de supercalculateurs plus grands et plus rapides. Les simulations du Dr Vrabec suivent maintenant les mouvements et interactions individuels de milliards voire de billions de molécules simultanément.
En 2019, l’équipe a même établi un record mondial du plus grand système moléculaire jamais simulé en dynamique moléculaire, avec 21 billions d’atomes. Pour l’ammoniac, les chercheurs ont réalisé des simulations de dynamique moléculaire et de Monte Carlo à l’aide du code ms2, étudiant cinq mélanges courants impliquant l’ammoniac en génie chimique.
Des données précieuses pour la recherche sur l’ammoniac
Pour chaque mélange, les simulations ont généré des données décrivant l’équilibre vapeur-liquide (VLE) sur une large gamme de températures et de pressions, jusqu’à 50 mégapascals (500 fois la pression ambiante). Bien que des données sur les mélanges d’ammoniac soient recueillies depuis plus d’un siècle, leur couverture reste étonnamment étroite en raison de l’effort expérimental prohibitif nécessaire.
L’analyse des données de simulation a montré que bien que l’ammoniac soit un composant de tous les systèmes étudiés, les graphiques de VLE résultants diffèrent considérablement selon les mélanges moléculaires. L’article et ses données supplémentaires offrent plus de 400 nouveaux points de données pour chaque mélange étudié, particulièrement précieux pour les conditions extrêmes difficiles à étudier.
En comparant leurs résultats avec d’autres ensembles de données VLE existants, les deux scientifiques ont identifié dans certains cas des divergences significatives avec les mesures et prédictions d’autres groupes de recherche, suggérant que des sources de données expérimentales spécifiques devraient être utilisées avec prudence.
À l’avenir, l’augmentation des performances de calcul haute performance pourrait permettre de simuler non seulement les propriétés mais aussi les processus thermodynamiques en utilisant des conditions aux limites proches des applications réelles. En attendant, les résultats de l’équipe démontrent la valeur de la dynamique moléculaire et de la simulation de Monte Carlo, fournissant une nouvelle compréhension du comportement de phase que les ingénieurs peuvent utiliser pour développer de nouvelles technologies basées sur l’ammoniac.
Légende illustration : Les différents mélanges de molécules présentent des propriétés thermodynamiques différentes, qui influencent le comportement des processus chimiques. Image : J. Chem. Eng. Data 2024, 69, 2, 573-589.
Article : « High-Pressure Fluid-Phase Equilibria and Henry’s Constants of Supercritical Gases in Ammonia » – DOI: 10.1021/acs.jced.3c00327